1. Introduction : Short peptide drug design—from “easily degradable” to “rock stable”
In the vast landscape of drug research and development, short peptide It is winning more and more attention with its unique "medium body". Compared with traditional small molecule drugs, short peptides can form richer hydrogen bonds and hydrophobic interactions, usually have higher binding specificity and affinity, and their metabolites are less toxic; compared with large molecule biopharmaceuticals such as antibodies, short peptides exhibit better tissue permeability, lower production costs, and weaker immunogenicity. It can be said that short peptides have found an attractive balance between "small" and "big".
However, natural short peptides (composed of L-amino acids) face a "natural shortcoming" - Protease degradation in vivo . Proteases in the human body can quickly recognize and cleave L-form peptide chains, resulting in the in vivo half-life of many short peptide candidates being only a few minutes to tens of minutes. This problem has long restricted the clinical transformation of short peptide drugs.
An elegant solution is to replace the L-amino acid with its chiral enantiomer - D-amino acid . D-amino acids are not the building blocks of natural proteins, so they can effectively escape recognition and degradation by proteases, extending the metabolic half-life of short peptides from minutes to hours or even days. Not only that, chiral flipping can also unlock the conformational space that is inaccessible to L-form peptides, making it possible to discover new binding modes. It is this "antichirality" design that makes D-amino acid short peptides one of the research hotspots in recent years.
Figure 1. Short peptide drug design process based on traditional models
2. Modern structural prediction tools : A new paradigm in the AI era
Traditionally, obtaining the structure of short peptide-protein complexes highly relies on experimental methods (such as NMR, X-ray crystallography) or molecular docking. The former is time-consuming and labor-intensive, while the accuracy of the latter is often unsatisfactory when faced with flexible short peptides. With AlphaFold3 and Chai-1 With the advent of the co-folding deep learning model, this situation has been completely rewritten.
These models are no longer limited to folding predictions of single proteins but can directly predict protein-ligand complex , covering a variety of ligand types such as small molecules, nucleic acids, and short peptides. For short peptide design, this means we can quickly obtain high-confidence binding mode hypotheses, compressing sequence-to-structure cycles from weeks to tens of minutes.
But you need to be sober-cautious . Two recent benchmark studies (the work of Lai Luhua's team Benchmarking co-folding methods to predict the structures of covalent protein–ligand complexes (https://doi.org/10.1038/s41401-025-01721-5), and the adversarial test published in *Nature Communications* Investigating whether deep learning models for co-folding learn the physics of protein-ligand interactions (https://doi.org/10.1038/s41467-025-63947-5)) systematically evaluates the physical plausibility of current mainstream cofolding models. The results show that although these models perform well on standard test sets, when faced with "adversarial" scenarios such as the binding site being destroyed by mutations, or the ligand's charge or hydrogen bonding ability being drastically changed, the models generally show a strong bias towards the original binding site - even if it should no longer bind physically, the model still tends to "stuff" the ligand back to its original place, and produce non-physical structures such as atomic overlap and steric conflict. Even more worryingly, high-confidence metrics such as pLDDT often fail to reflect these errors. In other words, the model may be "confidently" outputting a structure that is physically impossible.
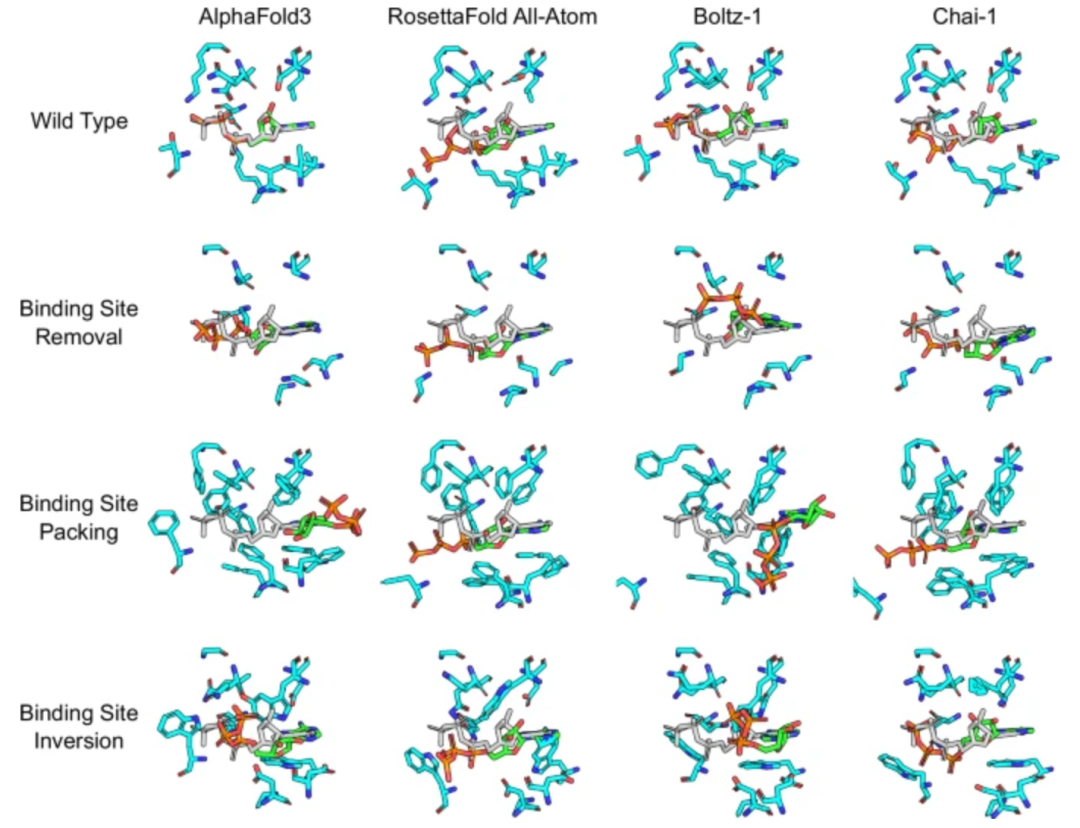
Figure 2. It is difficult for different deep learning methods to accurately predict the protein-ATP binding mode after pocket modification.
Therefore, researchers should regard AI predictions as “an efficient but occasionally erroneous preliminary screening tool” rather than a final answer. Physical verification - Especially molecular dynamics (MD) simulation - is a necessary link to make up for the lack of physical understanding of AI models. The following will introduce a complete set of "AI prediction + structure mapping + MD simulation" workflow, specially tailored for the design of D-amino acid short peptides.
3. “Prediction + Simulation” full process solution for D-amino acid short peptides
Most AI models (including AlphaFold3) treat D-amino acids as "non-standard small molecule ligands" instead of "protein residues" by default. This will cause subsequent MD simulations to face problems such as confusion in atomic naming and lack of standard force field topology. The automated assembly line we built has opened up " File preparation → AI prediction → Structural correction → MD simulation "The closed loop makes format compatibility no longer an obstacle.
Step One: Convert Sequence to SMILES—Precisely Control Chirality
For D-peptide, AI models usually require SMILES string The form is entered as "small molecule". The most critical step is to accurately label the chirality of each α-carbon. The workflow that can be considered achieves high-precision SMILES generation through the following logic:
-
LEaP build
-
Chirality flip
-
Topology transformation
-
automatic Chirality mark
# Example: Generate SMILES for D-His-D-Lys-D-His-D-Ala-D-Pro-D-Serpython scripts/d_peptide_smiles.py"NH2-D-His-D-Lys-D-His-D-Ala-D-Pro-D-Ser-Ac"-o ./prep_dir
Step 2: Automated structure prediction—one-click call
Define the target sequence and short peptide information through the JSON configuration file, and you can call AlphaFold3, Boltz-1 or Chai-1 for prediction with one click. It is recommended to perform multiple repeated predictions (with different random seeds) on the same system to evaluate structural convergence.
"targets" "name" "target" "chain" "A" "sequence""peptides" "name" "peptide" "chain" "B" "sequence" "NH2-D-His-D-Lys-D-His-D-Ala-D-Pro-D-Ser-Ac" "is_d_amino_acid" true"predictions" "method" "af3" "run_script" "./run_af3.sh"
Step 3: Structural correction and "re-peptidation" - get the format back in place
In the CIF/PDB file output by AI, D-peptide is usually labeled as "small molecule LIG", and the atom names (C1, N1...) and residue names do not comply with MD simulation standards at all (AMBER force field requires standard naming such as N, CA, C, O, etc.). we developed graph matching correction algorithm , the steps are as follows:
-
Topology aware
-
subplot isomorphism match
-
coordinate mapping
python scripts/map_af3_to_tleap.py -ttemplate.pdb -i predicted.cif -o corrected_peptide.pdb -c complex.pdbStep 4: Processing of modified amino acids and special groups
Many D-peptide designs introduce unnatural amino acids or chemical modifications (such as phosphorylation, methylation, PEGylation, fluorescent labeling, etc.). For these "non-standard" parts, standard AMBER force fields (such as ff14SB) cannot provide parameters directly. At this time it is necessary to adopt Hybrid modeling strategy :
-
Standard skeleton part : If the modifying group is attached to a standard amino acid side chain (for example, an acetyl group is attached to the side chain amino group of lysine), the amino acid body can be retained as a standard residue and only use the General Force Field (GAFF2) generation parameters for the extra group.
-
Completely unnatural amino acids : If the entire residue does not exist in the standard library, it needs to be disassembled into "similar standard residues + modification groups", or use GAFF2 to generate full parameters from scratch, and ensure compatibility with the surrounding environment through matching of atom type, charge and bond parameters.
-
connection handling :use
tleapofbondcommands or third-party tools such asacpype,antechamber) Covalently attach modifying groups to standard residues while checking the plausibility of valency and charge.
For specific operations, you can first use
antechamber
Generate the GAFF force field parameters of the modifying group, and then use
resp
or
gasteiger
method to calculate the charge, and finally pass
tleap
Splice the modifying group with the standard residue into a complete residue. This step requires careful checking of atom naming and connection order to avoid "broken bonds" or "misalignments." For common modifications (e.g. phosphorylated serine/threonine/tyrosine), the AMBER force field already provides ready-made parameters (e.g.
SER
phosphorylated version of
SEP
), can be called directly.
Step 5: Tool selection for molecular dynamics simulation and the significance of energy calculation
Simulation tool selection
The current mainstream molecular dynamics simulation software includes
AMBER
,
GROMACS
,
NAMD
,
OpenMM
wait. For short peptide-protein complex systems, it is recommended to give priority to
AMBER
(Rich force field parameters, especially suitable for protein-peptide systems) or
GROMACS
(Fast computing speed, active community). If complex custom residues or modified groups are involved, AMBER’s
tleap
Cooperate
parmchk2
and
antechamber
Provides a relatively complete solution. OpenMM is suitable for GPU acceleration and combination with machine learning potential functions. When choosing, you need to consider the team's familiarity, force field compatibility, and whether subsequent calculations of binding free energy are needed (for example, AMBER's MMPBSA.py is more mature). This project suggests that OpenMM is more appropriate, as it can support large-scale, automated simulation scenarios and facilitate peptide design and screening.
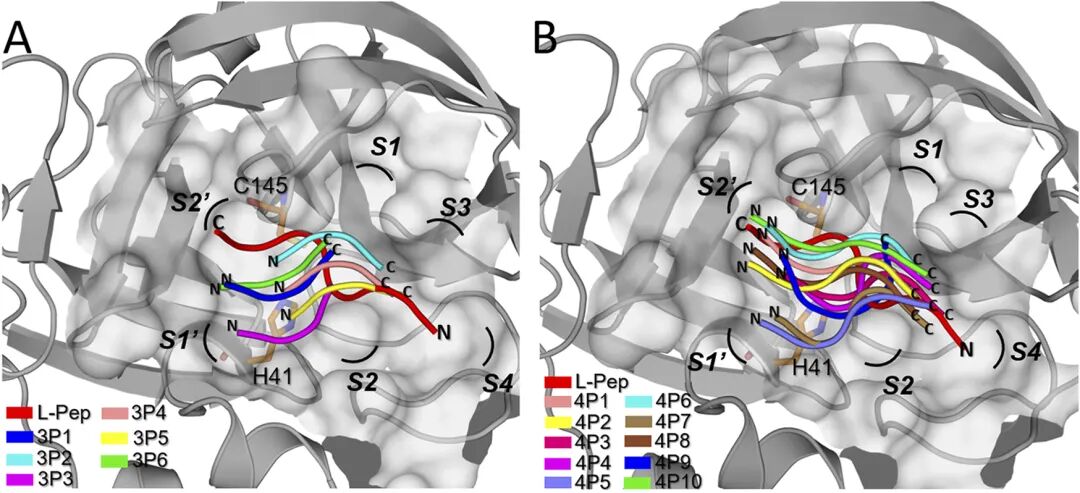
Figure 3. The binding mode of D-peptide and SARS-COV2 main protease obtained based on molecular East-West simulation
The meaning of energy calculation
Molecular dynamics simulations are more than just "watching animations." By analyzing the energy terms in the simulated trajectory, the merits of the design can be quantified:
-
Potential energy component analysis : Separate bond energy, angular energy, dihedral energy, van der Waals energy and electrostatic interaction energy to help determine the main driving force between the short peptide and the target.
-
Binding free energy (MMPBSA/MMGBSA) : This is the core metric for assessing affinity. By calculating the free energy difference between the protein-peptide complex and the individual protein or individual peptide, the binding free energy (ΔG) can be obtained. This method can relatively rank short peptides with different sequences and different modifications, and guide the optimization of lead compounds.
-
Residue contribution breakdown : Decompose the binding free energy into each residue and identify "hotspot residues" - the key positions that contribute the most to binding. These hot spots are often the focus of mutations or modifications.
-
entropy estimate : Estimate the binding entropy change through canonical pattern analysis or quasi-harmonic analysis to help understand the cost of conformational flexibility during the binding process.
It should be emphasized that the absolute value of the energy calculation is highly dependent on the force field parameters and simulation settings. relative comparison (For example, the ΔG difference between different short peptides under the same target) is more meaningful than the absolute value. It is recommended to always compare with positive control molecules under exactly the same computational conditions.
Multiple Strategies for Energy Calculation
MMPBSA/MMGBSA is a common starting point for evaluating short peptide-protein binding affinity. Because it has a relatively low computational cost and can provide residue energy decomposition, it has practical value in sequence screening. However, researchers should realize that MMPBSA is highly sensitive to the quality of conformational sampling, and its absolute energy value is greatly affected by force field parameters and dielectric constant settings, so it is more suitable for relative comparison (like ranking of different short peptide designs at a site) rather than as a reliable prediction of absolute affinity.
On the basis of MMPBSA, the following three other energy calculation strategies can be considered for different research scenarios:
① deep learning Affinity prediction—for high-throughput primary screening accelerator
In recent years, deep learning affinity prediction models for protein-peptide interactions have developed rapidly. For example, the GraphPep framework proposed by Huang Shengyou's team at Huazhong University of Science and Technology, by constructing a protein-peptide interaction graph and using a hierarchical graph neural network to model interface geometry and energy characteristics, significantly outperforms traditional scoring functions in binding energy prediction and conformation scoring tasks on multiple benchmark data sets. Similarly, the PepBAN framework uses protein language models and bilinear attention networks to effectively learn the local interaction patterns of peptide-protein pairs, and also shows prediction advantages in non-standard amino acid systems such as cyclic peptides. In addition, MMPepPro demonstrated good generalization performance by integrating macroscopic binding trends and microscopic residue interaction characteristics, and was trained on nearly 20,000 peptide-protein complexes.
The common advantage of these deep learning methods is that Very fast reasoning (milliseconds to seconds), suitable for rapid sorting of thousands of candidate short peptides by binding propensity. However, it should be noted that the prediction reliability of such models is highly dependent on the coverage of the training set - when the designed short peptides contain unnatural amino acids, D-form configurations or novel chemical modifications, the model may fail to extrapolate. Therefore, deep learning affinity prediction is more suitable as high-throughput screening the first course filter , and its output needs to be combined with physical simulation for subsequent verification. But these deep learning methods may suffer from overfitting risks, making it difficult for biocomputing experts to truly trust these tools.
② Absolute binding free energy calculation (ABFE) - "ab initio" evaluation without reference ligand
When there is no known active molecule as a reference for a target, the deep learning model cannot provide reliable relative ranking, and the absolute value of MMPBSA lacks credibility. at this time, Absolute Binding Free Energy calculation (Absolute Binding Free Energy, ABFE) Becoming a more rigorous choice. ABFE "decouples" the ligand from the binding pocket (gradually closing its interaction with the protein and solvent) by constructing a thermodynamic cycle, and calculates its free energy difference between the bound state and the free state. In recent years, the emergence of automated processes such as BindFlow has significantly lowered the threshold for using ABFE. This tool is based on the GROMACS engine, has built-in support for a variety of small molecule force fields such as GAFF and OpenFF, and has completed system verification on 139 ligand-target pairs. The consistency between its predictions and experiments is close to the gold standard method in the field.
The advantage of ABFE is that Does not rely on any reference ligand , which can directly give an estimate of the binding free energy of a single molecule and has a clear statistical mechanics basis. However, it is computationally expensive (usually requiring tens to hundreds of nanoseconds of enhanced sampling simulation) and is sensitive to the quality of the initial structure. For non-standard systems such as D-peptide, special attention needs to be paid to whether the field parameters cover the energy description of the non-natural chiral center. ABFE is particularly suitable for the following scenarios: designing the first short peptide for a new target, evaluating whether an existing design has nanomolar binding potential, or verifying whether the AI-predicted structure is thermodynamically feasible.
③ Relative free energy perturbation calculation (FEP) - precise optimization when there are positive molecules
The most common and practical scenario in drug discovery is: there is already a positive molecule (or initial hit molecule) with known activity, and it is necessary to evaluate the improvement or decrease in affinity after a series of modifications. at this time, Relative Binding Free Energy (RBFE/FEP) is the best choice. FEP is based on the principle of thermodynamic cycles and directly calculates the relative binding free energy (ΔΔG) by simulating the free energy difference between the bound and free states of two molecules with similar structures. The biggest advantage of this approach is that error cancellation ——Since molecule A and molecule B are highly similar in structure, the systematic errors tend to cancel each other out during the calculation process, so the prediction accuracy of FEP is usually significantly better than MMPBSA and ABFE.
In practice, FEP requires small chemical changes between ligand pairs (such as the replacement of a functional group or the addition of a methyl group), and the ability to construct a reasonable "alchemical path" to gradually "transform" from one molecule to another. When there are multiple molecules to be tested, the positive molecule is usually used as a reference node to construct a star topology or linear path for relative ranking of all ligands. FEP has been widely used in industrial drug discovery, and tools such as Schrödinger's FEP+ and open source QligFEP provide mature implementation solutions. For D-peptide optimization scenarios—such as site-by-site replacement of D-amino acids or adjustments to side chain modifications while maintaining the binding mode—FEP is an ideal tool to quantify the effect of the modification.
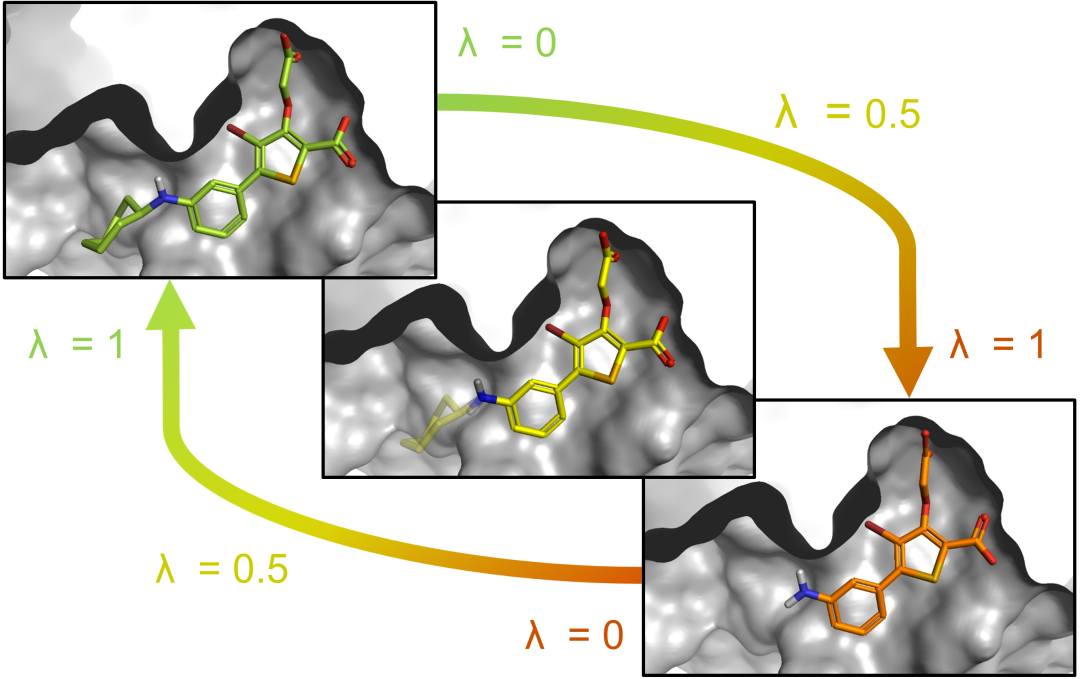
Figure 4. Schematic diagram of FEP relative free energy calculation
Strategy selection and considerations
The above three methods complement each other in terms of computational cost, accuracy and applicable scenarios: deep learning affinity prediction is most suitable for ultra-high-throughput primary screening (second level/thousands of items); ABFE is suitable for absolute affinity estimation without reference ligands (day level/single molecule); FEP is the gold standard when there are positive molecules (hour to day level/ligand pair). Researchers should make reasonable choices based on their own research stage and resource conditions. No matter which method is used, it is recommended to establish a closed-loop verification with experimental data: for systems with positive molecules, the calculation method should first be calibrated with this molecule (to ensure that FEP or ABFE can reproduce the experimental trend), and then extended to the molecules to be measured. At the same time, all energy calculation methods are limited by the accuracy and sampling adequacy of the force field, and their output should always be used as an aid to decision-making, not as a substitute for experiment.
4. In-depth analysis : How to evaluate your short peptide design?
Once a stable composite structure is obtained, the design quality can be quantified through multidimensional analysis:
A. Interface contact analysis
Use analysis scripts to automatically generate distance maps and contact maps between targets and short peptides. Distance maps provide a visual representation of how compact the binding pocket is; contact maps help identify key "hotspot" residues.
B. Kinetic stability analysis
-
RMSD : Evaluate the overall fluctuations of the complex during the simulation. A smooth curve means the binding pattern is stable.
-
RMSF : Identify which residues in short peptides are flexible and which are firmly anchored in the binding pocket.
C. Binding Free Energy Calculation (MMPBSA)
Based on the MMPBSA.py module of AmberTools, the free energy of the binding mode can be quantitatively calculated and predicted. This is a key indicator for evaluating the affinity of short peptides and can effectively distinguish the advantages and disadvantages of different designed sequences.
5. Limitations and important notes (please be sure to read it again and again)
Although the above process integrates the current state-of-the-art structure prediction and molecular simulation technology, we must frankly point out: There is no computational model or Automated process Able to be versatile and robust in industrial drug discovery scenarios . The following limitations and considerations need to be kept in mind by researchers in practice:
Modeling credibility assessment
-
Physical bias in AI predictions : As mentioned previously, cofolding models may over-memorize the binding patterns in the training data and ignore the true physicochemical constraints. therefore, The success of a design cannot be determined solely by the high-confidence scores output by AI. . It is recommended to always repeat predictions on the same system multiple times (at least 3-5 random seeds) to observe the convergence of the binding pattern. If different predictions give drastically different conformations, caution should be taken.
-
Molecular Dynamics Simulation force field limitations : Classical force fields (such as ff14SB, GAFF2) cannot describe complex behaviors such as chemical reactions, electronic polarization effects, and metal ion coordination. For systems containing metal ions, covalent inhibitors, or highly polarized groups, supplementary verification using QM/MM or polarization force fields may be required.
-
Undersampling problem : Conventional simulation times of 10-100 ns may not be able to fully sample all conformational changes of short peptides, especially for highly flexible peptides or systems with slow conformational transitions. If continuous RMSD drift or unstable binding mode is observed, the simulation time should be extended (at least 200-500 ns) or enhanced sampling methods (such as replica exchange, metadynamics) should be used.
-
Quality of modified amino acid parameters : When using GAFF2 to generate non-standard residue parameters, the charge calculation method (such as AM1-BCC, RESP) and atom type classification directly affect the reliability of energy calculation. Be sure to perform a visual inspection of the generated parameters : Are the bond lengths and bond angles reasonable? Does the charge distribution satisfy chemical intuition? If necessary, it can be compared and verified with the electrostatic potential calculated by DFT.
-
heavy reliance on expert knowledge : Every intermediate node in the entire process—from initial SMILES generation, AI predicted structure selection, to the quality of structure correction, setting of simulation parameters, and interpretation of trajectory analysis—requires the professional judgment of the researcher. There is no shortcut to "one-click generation" . It is recommended that the team have at least one member with molecular modeling and simulation experience responsible for quality control and anomaly identification of critical nodes.
-
Positive molecules serve as "Dinghai magic needle" : If there are positive molecules with known activity against the same target (such as reported active short peptides or small molecule inhibitors), It is strongly recommended to perform the exact same calculation process for this positive molecule . Its binding mode (whether the experimental conformation is reproduced), stability (whether the RMSD curve is stable) and energy (whether ΔG is consistent with the activity trend) are used as reference standards. Only when the calculation results of the design to be tested are significantly better than or equivalent to the positive molecule and the binding mode is reasonable, will it be worthy of further experimental verification. On the contrary, if the positive molecule itself performs poorly in the calculation (for example, the predicted conformation is significantly different from the experimental one, or it falls off during the simulation), it means that there is a problem with the current calculation settings, and the parameters need to be re-optimized or the simulation method needs to be changed.
-
Experimental feedback is the final judge : The output of a computational model is always just hypothesis . Any promising short peptide design should be verified by experiments (such as SPR, ITC, fluorescence polarization, cell viability assay). Iterative optimization of calculations and experiments is the right way to design drugs.
Applicable Boundaries and Misuse Risks of Energy Calculation Methods
It must be admitted that currently no energy calculation method can stably output reliable affinity rankings in all scenarios. The biggest risk of deep learning affinity prediction models is training set bias - when the designed short peptide is different in sequence or structure from the training data distribution (such as the introduction of D-amino acids, non-natural residues or cyclic structures), the prediction results of the model may be completely unreliable. Although the ABFE and FEP methods have a more solid foundation in statistical mechanics, their accuracy is highly dependent on whether the simulation time is sufficient (inadequate sampling will lead to biased free energy estimates), whether the force field parameters are accurate (especially for the charge and dihedral angle parameters of non-standard residues), and whether there are hidden conformational changes that are not sampled. In addition, the FEP method requires clear chemical similarity between ligand pairs (usually Tanimoto similarity coefficient >0.7), otherwise the “alchemical path” will lose physical meaning. Researchers should fully understand the respective assumptions and failure modes of these methods and avoid over-interpreting calculation results as "experimentally determined affinities". When the conclusions given by different methods are inconsistent, methods with a more solid physical foundation are preferred (FEP > ABFE > MM(PB)GBSA > Deep Learning), but experimental verification is still required as the final standard.
6. Summary
The set introduced in this article AI prediction + structural mapping + MD simulation The workflow aims to solve the core technical bottlenecks in D-amino acid short peptide drug design - from chiral SMILES generation, AI structure prediction, to format correction, modification group parameter construction and physical simulation verification. This process takes advantage of the high-throughput screening capabilities of AI and ensures physical plausibility through molecular dynamics simulations based on classical force fields, while emphasizing reliance on positive controls and expert knowledge.
However, we emphasize again: Calculation is always an auxiliary, experiment is the standard . Today, with the rapid development of short peptide drug research and development, tool innovation continues to expand the boundaries of design, but only by combining careful calculations with solid experiments can we go further. Welcome to have an in-depth discussion with us and start your journey of D-peptide design.
