Have you ever wondered why the enzymes in washing powder remain active in hot water? Why don't some expensive biologic drugs need to be kept in the refrigerator? Behind all this, there is a key word - protein thermal stability. Making proteins "heat-resistant" is a core task in industry, medicine and scientific research. A heat-labile enzyme may "die" before it can take effect; an unstable antibody drug may become ineffective during transportation.
So, how do scientists "wear insulating suits" on proteins so that they can survive high temperatures? Today, let’s talk about the various “weapons” and “tactics” in this “protein heat resistance upgrade war”.
Chapter 1 :Old tactic——Rational Design
If protein is compared to an exquisite building, rational design is to take the architectural drawings and accurately reinforce or adjust certain key nodes. This requires us to have a thorough understanding of the three-dimensional structure of proteins.
-
“Adding a Lock”: Introducing Disulfide Bonds Imagine adding steel cables to certain parts of a building to lock the loose parts tightly. In proteins, we can introduce cysteine at two spatially close sites. They will form a pair of "disulfide bonds", like a molecular lock, making the protein structure more "rigid" and less likely to be dismantled by high temperatures.
-
"Reducing rocking": reducing conformational entropy (Entropy Reduction) The flexible region of a protein, like a willow branch in the wind, sways unsteadily and is most likely to "fall apart" at high temperatures. what to do? Put a "cast" on it!
-
Proline strategy (Proline): Proline is a rigid amino acid with its own "loop". Placing it in a flexible area is like attaching a fixed bracket to a swinging willow branch, greatly reducing its space for movement.
-
Glycine replacement (Glycine): Glycine is the most "flexible" amino acid. Replacing it with a chiral amino acid (such as alanine) is equivalent to replacing a soft rope with a hardwood stick, and the skeleton suddenly becomes harder.
-
"Filling the gaps": optimizing the hydrophobic core (Hydrophobic Core) If there are holes (cavities) inside the protein, it is like bubbles in the load-bearing walls of a building, which will easily collapse when heated. Rational design will use mutations to fill these holes with larger or more suitable amino acids, thereby enhancing the internal van der Waals forces and hydrophobic interactions, making the internal structure denser.
-
"Stretching the network": Optimizing surface charges and ionic bonds (Salt Bridges) Rearranging charges on the protein surface and introducing "salt bridges" (ionic bonds) is like adding a reinforcement network to the outside of a building to make the local structure more compact and stable.
-
“Looking for weaknesses”: B-factor Directed Rigidification B-factor can be understood as the “thermometer” or “sway index” of each atom in the protein structure. The higher the index, the more "moving" that place is, that is, the more fragile it is. Rational design specifically targets these "soft spots" and makes them hard through mutations.
Chapter 2 : The way to be smart - semi-rational design (Semi-rational Design)
Making changes based on experience is sometimes like finding a needle in a haystack. Semi-rational design uses evolutionary information or computational predictions to first narrow the "battlefield" and then conduct experimental verification, which is more efficient.
-
"Take the majority": Consensus Design This is a very interesting idea: line up sequences from different species in the same protein family and see which position and which amino acid appear most. If evolution thinks this "consensus residue" is good, then it will most likely contribute to stability. When we change the corresponding site of the target protein to it, the stability can often be improved.
-
“Tracing back to the source”: Ancestral Sequence Reconstruction (ASR) This is a technology that “travels” back to ancient times. Scientists use algorithms to predict what the ancestor of a certain protein looked like tens or even hundreds of millions of years ago. It turns out that these "ancestral" proteins are often more heat-resistant than today's "descendants"! This gives us an excellent design blueprint.
Chapter 3 :AI Pilot——Computational Design and Deep Learning (Computational & AI Methods)
This is the most cutting-edge and exciting field right now! We no longer rely solely on experience and simple rules, but let AI and powerful computing software help us predict and design.
(1) Force field-driven “prophet”
This type of software is based on physical and chemical principles and guides design by calculating "whether the mutated protein is more stable or less stable" (ΔΔG).
-
FoldX: extremely fast scanner
-
Features: Fast! It only takes a few seconds to calculate a single point mutation, and it can quickly scan all sites of a protein to find potential stabilizing mutations.
-
Suitable for: large-scale preliminary screening, quickly locking targets.
-
Official website: https://foldxsuite.crg.eu/
-
Rosetta: All-around “Design Master”
-
Features: Extremely powerful and highly accurate, it is the "Swiss Army Knife" in the field of protein design. its
ddG_monomerThe protocol is one of the gold standards for calculating stability changes, and can also be used for de novo design, structural prediction, etc. -
Suitable for: pursuing high-precision and complex design tasks.
-
Disadvantages: steep learning curve and large amount of calculation.
-
Official website: https://www.rosettacommons.org/
-
Other fast prediction tools (PoPMuSiC / CUPSAT)
-
These are lightweight online tools based on statistical potential energy. They are simple to operate. You can quickly get results by inputting the PDB structure. They are also very suitable for preliminary screening.
-
PoPMuSiC official website: https://babylone.ulb.ac.be/popmusic/
-
CUPSAT official website: http://cupsat.tu-bs.de/
(2) AI-driven “Magician”
ProteinMPNN
This is a generative AI model. Give it a protein skeleton structure, and it can recommend the most stable and natural amino acid sequence like an AI painting. The effect is stunning and it is currently one of the preferred tools for sequence design.
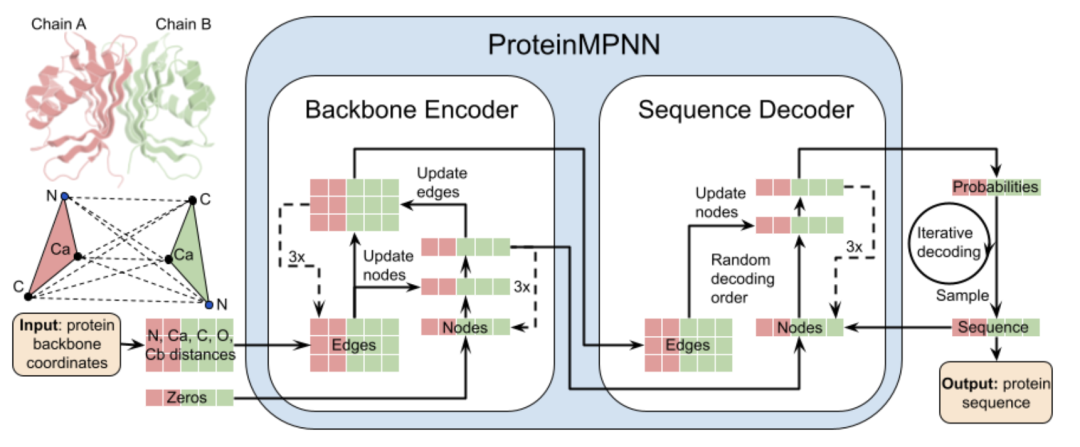
It is difficult for traditional computers to understand three-dimensional structures, but ProteinMPNN translates the structure diagram into a mathematical language it can understand:
encoder
-
Turn the structure into a "relationship graph": it treats each amino acid residue as a node, and the spatial proximity relationship (such as distance, angle) between residues as edges, transforming the entire protein structure into a topological graph.
-
Multi-dimensional "perception" information: The model not only records nodes, but also extracts rich edge features (such as the distance between Cα atoms, the main chain dihedral angle, and even the all-atom distance of Cβ virtual atoms), allowing the model to "see" the stereochemical constraints between residues.
-
"Message passing" and "attention": Through the multi-layer graph neural network, each node will continuously collect information about neighbor nodes (message passing), and judge the "importance" of different neighbors through the SE3-equivariant attention mechanism. This process allows the model to understand the global topology of the entire protein, rather than looking at individual sites in isolation.
decoder
After "reading" the structure, the model begins to generate sequences. The way it is written is unique:
-
"Fill in the blanks" in random order: Unlike traditional models that write from left to right, ProteinMPNN randomly shuffles the decoding order during training. This is equivalent to letting it start writing from any position, which can better capture the long-range dependencies between amino acids and avoid relying only on adjacent residues.
-
Autoregressive generation: When generating, the model determines amino acids site by site. When deciding the next site, it not only looks at the backbone structure, but also refers to other sites that have been generated (just like when filling in the blanks, combined with context)
Training secrets - why is it so "stable"?
Its success is also due to two clever designs:
-
Adding "noise" training: Gaussian noise is deliberately added to the input skeleton coordinates during training. This forces the model not to be obsessed with small errors in local atomic positions, but to learn to pay attention to the overall topological characteristics (such as the distribution pattern of the hydrophobic core and polar surface). Therefore, it has strong generalization ability and can design sequences with a very high experimental success rate.
-
Multi-chain and symmetry awareness: The model can distinguish intra-chain and inter-chain interactions. When dealing with multimers or symmetric proteins, it can ensure that the same amino acids are designed at symmetric positions to ensure correct assembly of the structure.
AlphaFold Series
AlphaFold series: AlphaFold2 not only predicts structures, its prediction confidence score (pLDDT) itself is a "stability indicator". Places with higher scores generally have more stable structures. There are also new methods using AlphaFold to assist in the design of stable mutations.
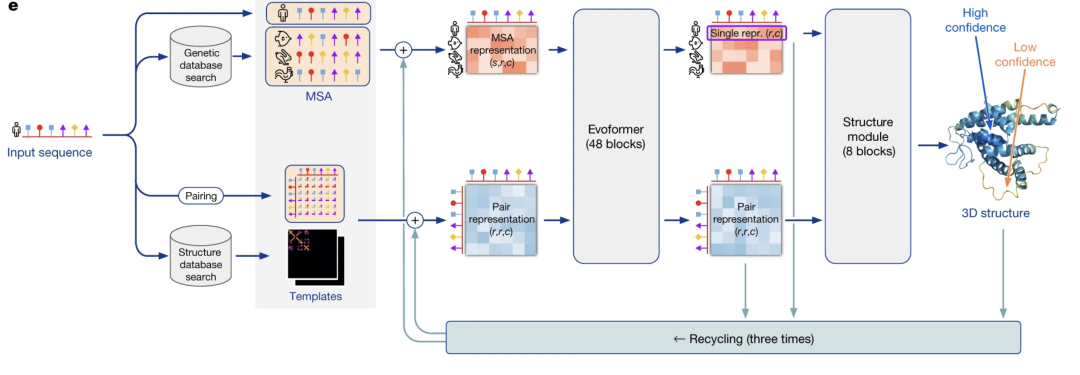
When AlphaFold2 predicts the protein structure, it will output a pLDDT score (0-100) for each amino acid, indicating the confidence of the prediction at that position.
| pLDD | meaning |
| >90 | The structure is very definite, usually meaning the area is rigid and stable |
| <70 | The structure is uncertain and often corresponds to flexible regions or disordered regions. |
The relationship between pLDDT and stability: high-scoring area = rigid structure = tends to be thermally stable; low-scoring area = flexible = often the "weak link" of the protein.
How to use it to assist design?
-
Identify unstable areas: Areas with low pLDDT (especially the loop area) are targets that need to be "reinforced"
-
Screen design candidates: After generating mutants using tools such as ProteinMPNN, use AlphaFold2 to predict the structure, and only retain mutants with high average pLDDT for experiments.
-
Guided mutation strategy: Target the low pLDDT region and introduce rigid modifications such as proline and disulfide bonds
Chapter 4 : How to judge? ——Evaluation indicators and experimental verification
What I have learned on paper is ultimately shallow, and I know that I have to do it in detail. No matter how good the design is, it must ultimately be verified by experiments.
-
Melting temperature (Tm): This is the most commonly used indicator and refers to the temperature at which 50% of the protein "melts" (unfolds). The higher the Tm value, the more heat-resistant the protein is.
-
Half-life (t1/2): The time required for a protein to remain half active at a specific elevated temperature. It measures kinetic stability.
-
T50: The temperature required for the protein to retain 50% activity after heating for a period of time.
Follow us and witness a new era of computational changes in protein research and development!
Contact information: information@yunfeidu.com
👇 Click on the business card below to follow "Yunfeidu Technology"
